In this quick tutorial, we shall run SaVor to generate a union set of structural variants from a small set of Arabidopsis whole genome samples from NCBI.
For brevity, we shall use BAM files, their associated indices of 4 Arabidopsis thaliana and the TAIR10 reference sequence contained in the SaVor tutorial Google Drive directory.
This tutorial assumes you have;
Set up and configured your snakemake mamba/conda environment and verified snakemake is installed and functional. For instructions, please refer to the prerequisites here
You have cloned the repo onto your local machine.
Step 0: Download Reference Sequence and BAM Files.¶
Create a new directory and name it REFERENCE. Download the TAIR10 reference genome from this link and move it into the REFERENCE directory. Additionally, create an empty directory named bams/ and download all files from this Google Drive folder into it.
Step 1: Setup the workflow config.yaml¶
Change into the config directory and edit the following options in the config.yaml file with a text editor of your choice.
samples:- Edit this path to point to thetest.csvsample sheet.
include_contigs:- Edit this path to point to the fileinclude_contigs.csvcontaining the contig/chromosome IDs of the TAIR10 Reference.
While in the config directory, create an empty file with all the contig IDs of your reference genome. Below are the contig IDs for the TAIR10 reference you downloaded earlier.
Chr1
Chr2
Chr3
Chr4
Chr5Alternatively, you can easily determine the contig IDs from the reference file itself using the grep command.
grep "^>" ../REFERENCE/TAIR10_REF.fna > include_contigs.csvsv_merge- Change the value to1to generate a union set of SVs of the 4 samples.
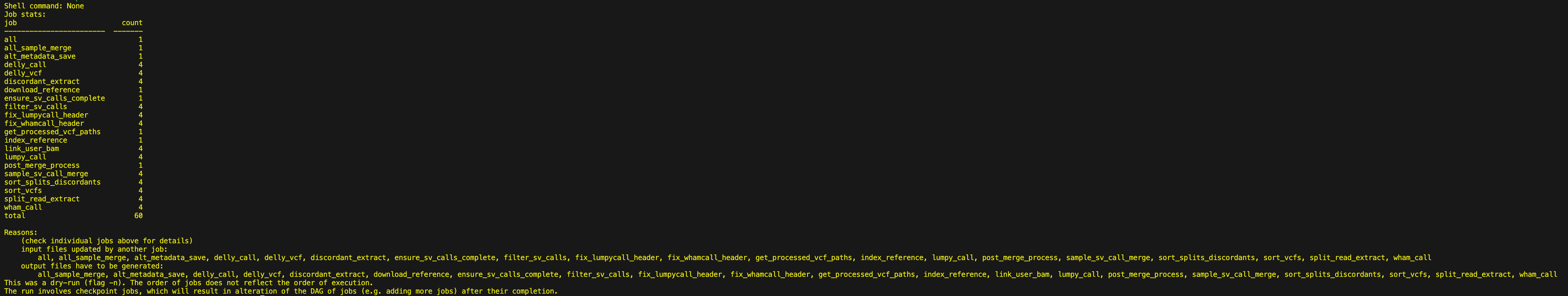
Step 3: Perform a Dry Run¶
Change out of the config directory and into the SaVor root directory. Next, activate the snakemake conda/mamba environment unless its already active. Run the following Snakemake command to generate a list of jobs SaVor will execute. This should execute in a couple of seconds. If you encounter any errors, ensure you are in the repository root directory and verify whether you have all necessary configuration and data files present.
snakemake --cores 1 -np --workflow-profile workflow-profiles/default/If your workflow is configured correctly, you should see the following message print out.
Step 4: Perform a Wet Run¶
Once your dry run completes successfully, you may remove the n option above to run the workflow.
# This assumes you have at least 8 cores on your local machines CPU.
snakemake --cores 8 -np --workflow-profile workflow-profiles/default/